
W okresie świątecznym, pod koniec grudnia 2019 roku, fundacja Siepomaga ogłosiła wspaniałą wiadomość: dotyczyła ona pozytywnego zakończeniu największej zbiórki w historii działania fundacji. Fundusze zbierano dla kilkumiesięcznego Alexa, u którego zdiagnozowano rdzeniowy zanik mięśni (ang. Spinal Muscular Atrophy – SMA). Wysiłek, jakiego podjęli się rodzice chłopca oraz pomoc udzielona przez tysiące osób, dały w efekcie możliwość podjęcia walki z tą śmiertelnie niebezpieczną chorobą.
Czym jednak jest rdzeniowy zanik mięśni i co jest przyczyną tej choroby? Jakie są obecnie dostępne metody leczenia? Czy jej leczenie jest tak horrendalnie drogie?
Rdzeniowy Zanik Mięśni (ang. Spinal Muscular Atrophy – SMA) i jego typy
Rdzeniowy zanik mięśni (SMA) jest rzadką monogenową chorobą genetyczną, charakteryzującą się postępującym osłabianiem i stopniowym zanikiem mięśni szkieletowych, co jest spowodowane obumieraniem neuronów ruchowych odpowiadających za kontrolę pracy mięśni [1]. Jeszcze do niedawna wyznacznikiem typu SMA był wiek, w którym zaobserwowano pierwsze objawy choroby u pacjenta. W ten sposób wyróżniało się cztery typy choroby [2,3]. Obecnie coraz częściej stosuje się jednak klasyfikację opartą na najwyższym osiągniętym etapie rozwoju ruchowego (samodzielne siedzenie, stanie z podparciem, stanie bez podparcia).
Najczęściej występującą formą rdzeniowego zaniku mięśni jest typ 1 (SMA1) zwany również chorobą Werdinga-Hoffmana. Wstępne objawy uwidaczniają się nagle w pierwszych tygodniach lub miesiącach życia, dziecko nie nabiera umiejętności samodzielnego siedzenia. Stan zdrowia niemowląt pogarsza się gwałtownie – mięśnie tułowia, kończyn czy przełyku ulegają szybkiemu osłabieniu, co powoduje, że dzieci te mają osłabioną zdolność ssania, połykania oraz oddychania. Większość nie jest w stanie unieść główki oraz siedzieć bez podparcia. Pojawiają się również problemy z przełykaniem, które prowadzą do trudności z odżywianiem i spowolnionego wzrostu. U dzieci cierpiących na ten typ choroby pojawiają się również poważne problemy z oddychaniem spowodowane słabością mięśni oddechowych. Większość umiera przed ukończeniem drugiego roku życia. SMA1 jest najczęstszą genetycznie uwarunkowaną przyczyną śmierci niemowląt i małych dzieci [4].
Rdzeniowy zanik mięśni typu 2 (SMA2) charakteryzuje się osłabieniem mięśni u dzieci w wieku pomiędzy 6 a 18 miesiącem życia. Dzieci dotknięte chorobą tego typu potrafią siedzieć bez podparcia, niemniej jednak wraz z nasileniem objawów w późniejszym okresie życia umiejętność ta zanika. Osoby cierpiące na ten typ rdzeniowego zaniku mięśni nie są również w stanie stać oraz chodzić samodzielnie, w związku z tym potrzebują specjalistycznej opieki
i wsparcia. Ryzyko śmierci jest jednak znacznie niższe niż w przypadku SMA1.
Typ 3 rdzeniowego zaniku mięśni (SMA3), często nazywany chorobą Kugelberga-Welander, powoduje osłabienie mięśni zazwyczaj już po upływie wczesnego dzieciństwa. Dotyczy on chorych, którzy potrafili samodzielnie chodzić, jednak z upływem czasu i postępem choroby staje się to coraz trudniejsze, a nawet niemożliwe. Osoby z tym typem SMA z reguły prowadzą stosunkowo niezależne życie.
Wyróżnia się również typ 4 rdzeniowego zaniku mięśni (SMA4). W jego przypadku pierwsze objawy występują w wieku dorosłym, a osoby dotknięte tym typem choroby zazwyczaj doświadczają osłabienia mięśni prowadzącego do niedowładu nóg.
Rdzeniowy zanik mięśni dotyka średnio 1 na 8 – 10 tysięcy osób. SMA typu 1 jest zdecydowanie najczęstszym typem tej choroby i stanowi około połowę liczby wszystkich zachorowań. Typy 2 i 3 są rzadsze, natomiast SMA typu 4 występuje sporadycznie [5].
Genetyczne podstawy rdzeniowego zaniku mięśni
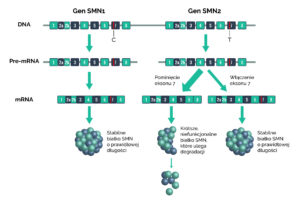
Rdzeniowy zanik mięśni występuje na skutek niedoboru białka SMN (ang. survival of motor neuron – „białko życia neuronów ruchowych”) w komórkach nerwowych. Jest ono niezbędne do prawidłowego funkcjonowania neuronów ruchowych. Instrukcje dotyczące budowy tego białka zapisane są w dwóch genach znajdujących się na 5 chromosomie – SMN1 i SMN2 [6]. Zdecydowana większość funkcjonalnego białka jest produkowana z genu SMN1. Gen SMN2 różni się od SMN1 w obrębie sekwencji kodującej zaledwie dwoma nukleotydami. Mimo, że różnica w sekwencji genów jest tak mała, odgrywa ona kluczową rolę w produkcji białka.
W konsekwencji tej zmiany znaczna większość białka produkowana z genu SMN2 jest krótsza, niefunkcjonalna i ulega szybkiej degradacji. Tylko ok 10-15% białka SMN produkowanego z genu SMN2 jest prawidłowe [7].
SMA spowodowane jest albo delecją genu SMN1 (93% przypadków) albo mutacją w jego obrębie (7%) [6]. Brak SMN1 może być częściowo zrekompensowana przez gen SMN2, jednak ilość funkcjonalnego białka SMN produkowanego z tego genu jest zbyt nikła, aby zapewnić prawidłowe funkcjonowanie komórek transmitujących sygnał nerwowy od mózgu do mięśni [7].
Ludzie różnią się między sobą liczbą kopii genu SMN2, co przyczynia się do występowania różnych typów choroby. Osoby posiadające większą liczbę kopii genu SMN2 produkują więcej funkcjonalnego białka SMN w komórkach nerwowych, a więc cechują się łagodniejszym przebiegiem choroby. U takich osób objawy choroby pojawiają się później i nie są tak dotkliwe, jak u osób z jedną lub dwiema kopiami tego genu [7].
Rdzeniowy zanik mięśni jest dziedziczony w sposób autosomalny recesywny. Oznacza to,
że schorzenie objawia się, tylko wtedy, gdy u danej osoby obie kopie genu SMN1 zawierają patogenną zmianę genetyczną. Rodzice takiej osoby nie mają żadnych objawów choroby i nazywani są w genetyce nosicielami – każdy z nich posiada jedną funkcjonalną kopię genu, a drugą uszkodzoną. Częstotliwość nosicieli w populacji europejskiej wynosi ok 1: 35-50. Osoby, które odziedziczyły zmutowany gen tylko od jednego rodzica, również nie mają objawów – w takiej sytuacji oni także są nosicielami. Prawdopodobieństwo odziedziczenia przez dziecko obydwu uszkodzonych genów od rodziców będących nosicielami wynosi 25%. Mechanizm ten nie zależy od płci dziecka, co oznacza, że choroba dotyka w takim samym stopniu zarówno chłopców, jak i dziewczynki [1]. Istnieje również ryzyko powstania u dziecka mutacji de novo, czyli zmian genetycznych w DNA potomstwa, niezależnych od rodziców, które pojawiają się spontanicznie.
Leczenie rdzeniowego zaniku mięśni
Rok 1995 był przełomowym momentem w historii rdzeniowego zaniku mięśni. To właśnie wtedy zespół prowadzony przez Judith Melki z Francji zidentyfikował gen odpowiedzialny za powstanie SMA [6]. Odkrycie to zmobilizowało organizacje zrzeszające rodziny z rdzeniowym zanikiem mięśni do zainicjowania zbiórki funduszy i w niedługim czasie możliwe było rozpoczęcie badań nad lekiem pozwalającym walczyć z tą śmiertelną chorobą.
Programy badawcze prowadzone przez pracownie uniwersyteckie i firmy biotechnologiczne szybko zaowocowały szeregiem obiecujących odkryć. Część z nich otrzymała kolejne dofinansowania, które pozwoliły na rozpoczęcie programów przedklinicznych (kolejny etap tworzenia leku), polegających na badaniu wpływu danego środka na zwierzęta laboratoryjne. Nieliczne z tych początkowych odkryć pomyślnie dotarły do etapu badań klinicznych z udziałem ludzi. Badania na tym etapie wzbudziły zainteresowanie dużych firm farmaceutycznych, gotowych zainwestować w najbardziej obiecujące programy.
Prowadzone latami badania zakończyły się sukcesem w 2016 roku. To właśnie w grudniu tego roku, w USA został dopuszczony pierwszy lek dla osób chorujących na rdzeniowy zanik mięśni.
Był nim związek zwany nusinersenem o handlowej nazwie Spinraza® [8].
Nusinersen
Nusinersen jest antysensownym oligonukleotydem, czyli syntetycznym, krótkim fragmentem łańcucha DNA, który jest komplementarny (pasujący) do konkretnego miejsca w cząsteczce RNA. Oligonukleotyd po podaniu wnika do jądra komórkowego komórek nerwowych, m. in. neuronów ruchowych wpływając na produkcję białka SMN z genu SMN2. Powoduje to zwiększenie ilości funkcjonalnego białka SMN powstającego z tego genu. W ten sposób gen SMN2 przejmuje funkcję uszkodzonego genu SMN1. Podwyższony poziom białka zapobiega obumieraniu neuronów motorycznych, co w połączniu z ciągłą rehabilitacją zazwyczaj przynosi poprawę stanu zdrowia chorego [9]. Znane są historie dzieci, które po kilku latach przyjmowania leku potrafią samodzielnie siedzieć, stać, a nawet chodzić. Te dzieci miały szczęście otrzymać lek jeszcze przed wystąpieniem objawów choroby, dzięki diagnozie na podstawie testów genetycznych.
W organizmie każdego człowieka niezwykle ważną funkcję odgrywa tak zwana bariera krew-mózg. Jest to bardzo „szczelna” struktura komórek, ściśle ze sobą połączonych za pomocą białek. Jej zadaniem jest izolacja układu nerwowego od wpływu środowiska zewnętrznego i stanowi ona naturalną przeszkodę w transporcie wielu substancji z krwi do mózgu. Ze względu na swój rozmiar cząsteczka nusinersenu nie jest w stanie przekroczyć bariery krew-mózg, co generuje potrzebę dostarczenia leku bezpośrednio do rdzenia kręgowego. Dlatego właśnie Spinrazę® podaje się dokanałowo, poprzez punkcję lędźwiową (wlew dooponowy). Zgodnie z zaleceniami i wskazaniami rejestracyjnymi roztwór do wstrzykiwań w fiolkach po 12 mg substancji podawany jest pacjentowi w następującym cyklu:
– pierwsza dawka podana jest pacjentowi możliwie jak najwcześniej po rozpoznaniu SMA;
– trzy kolejne dawki po upływie 2, 4 oraz 9 tygodni od pierwszego podania leku;
– kolejne dawki podtrzymujące podawane są regularnie co 4 miesiące aż do końca życia [10].
Nusinersen został dopuszczony przez amerykańską Agencję ds. Leków i Żywności (ang. Food and Drug Administration – FDA) na rynek amerykański dokładnie 23 grudnia 2016 roku. Z kolei pół roku później – 30 maja 2017 roku – Komisja Europejska dopuściła ten lek na terenie Unii Europejskiej do stosowania u wszystkich pacjentów cierpiących na SMA [11].
W Polsce od 1 stycznia 2019 roku leczenie nusinersenem jest objęte całkowitą refundacją
i dotyczy wszystkich osób cierpiących na SMA – niezależnie od typu choroby czy wieku [12]. Jest to bardzo ważna informacja dla pacjentów, ponieważ leczenie Spinrazą® jest niezwykle drogie. Koszt leczenia w pierwszym roku podawania leku wynosi ponad 700 tysięcy dolarów, natomiast kolejne dawki podtrzymujące to wydatek średnio ponad 350 tysięcy dolarów rocznie [13].
Terapia genowa
Zupełnie odmienną strategię walki z rdzeniowym zanikiem mięśni obrali twórcy innego preparatu, jakim jest AVXS-101, o zdecydowanie bardziej melodyjnie brzmiącej nazwie handlowej, jaką jest Zolgensma®. Jest to pierwsza i jedyna forma terapii genowej do stosowania u chorych cierpiących na SMA, która pomyślnie przeszła badania kliniczne i została dopuszczona do stosowania jako lek, jednak tylko na terenie Stanów Zjednoczonych i tylko dla chorych poniżej drugiego roku życia [14]. Terapia ta polega na leczeniu poprzez wprowadzenie do komórek pacjenta kwasu nukleinowego takiego jak DNA lub RNA, który wywiera efekt terapeutyczny zastępując uszkodzony i niefunkcjonalny gen. FDA dopuściła ten lek na rynek amerykański dokładnie 24 maja 2019 roku, natomiast w Unii Europejskiej i Japonii procedury związane z zezwoleniem na stosowanie AVXS-101 nadal trwają (stan aktualny w styczniu 2020 roku).
Zolgensma® to preparat zawierający biliony wirusów z rodziny AAV9, z których każdy przeszedł modyfikację genetyczną, w wyniku której stały się nośnikami transgenu SMN1 kodującego ludzkie białko SMN. Po podaniu do organizmu chorego, wirusy „dostarczają” komórkom pacjenta (w tym komórkom neuronów ruchowych) DNA z prawidłowym genem SMN1. Komórki, kiedy posiadają już odpowiednią instrukcję budowy brakującego białka, rozpoczynają jego produkcję. Wirusy nie zmieniają sekwencji DNA pacjenta, nie naprawiają również uszkodzonego genu SMN1, natomiast dostarczają do organizmy chorego jego syntetyczną kopię [15].
Podanie leku AVXS-101 możliwe jest tylko jeden raz w życiu, ponieważ po pierwszym kontakcie z wirusem, który przenosi syntetyczny gen SMN, pacjent nabywa trwałej odporności skierowanej przeciwko temu wirusowi. Twórcy leku Zolgensma® zakładają jednak, że jednorazowe podanie wirusa przyniesie dożywotni efekt terapeutyczny [16]. Niestety, dzisiaj nie jesteśmy w stanie stwierdzić, czy tak będzie w rzeczywistości, ponieważ dostępne dane obejmują okres nie dłuższy niż 5 lat. Lek podawany jest we wczesnym dzieciństwie, kiedy organizm intensywnie rośnie, czyli jego komórki się dzielą. Przy każdym podziale komórkowym dawka wirusa z prawidłowym genem ulega rozcieńczeniu. Znane są jednak historie dzieci, które po kilku latach po przyjęciu leku potrafią samodzielnie siedzieć, stać, a nawet chodzić. Te dzieci miały szczęście otrzymać terapię genową przed wystąpieniem objawów choroby. Zostały zdiagnozowane na podstawie testów genetycznych. Bez terapii osiągnięcie tych etapów rozwoju byłoby niemożliwe.
Niezwykłość wirusa używanego w leku Zolgensma® polega na jego zdolności do infekcji wielu tkanek oraz przekraczania bariery krew-mózg. To właśnie ta cecha wektora wirusowego umożliwia podanie leku w formie dożylnego zastrzyku [16]. Wcześniejsze próby terapii genowych w chorobach układu nerwowego najczęściej wiązały się z potrzebą dostarczania wirusa przenoszącego transgen bezpośrednio do układu nerwowego poprzez otwory wywiercone (sic!) w czaszce pacjenta.
Lek Zolgensma® jest obecnie najdroższym lekiem na świecie. Jednorazowe podanie wirusów zawierającego gen SMN wiąże się z kosztem wynoszącym ponad 2 miliony dolarów amerykańskich [17]. Cena terapii wzbudza skrajne emocje w związku ze swoją astronomiczną ceną. Novartis – konsorcjum, które jest właścicielem AveXis (firmy, która wymyśliła lek Zolgensma) – broni się, że cena nie jest zbyt wysoka, jeżeli porównamy ją z kosztem terapii za pomocą zatwierdzonego wcześniej nusinersenu [17]. Niestety nie jesteśmy w stanie przewidzieć, czy w przyszłości cena leku ulegnie obniżeniu i czy po dopuszczeniu leku w Europie będzie on refundowany podobnie jak Spinraza. Jeszcze do niedawna jedyną nadzieją dla chorych dzieci i ich rodzin na skorzystanie z terapii lekiem Zolgensma były zbiórki tak ogromnych kwot jak 9 milionów złotych, organizowane przez fundacje. Tylko wtedy, gdy zbiórka kończyła się sukcesem, możliwy stawał się wyjazd do Stanów Zjednoczonych, gdzie można otrzymać lek, tak jak to się zadziało w przypadku małego Alexa. Na przełomie stycznia i lutego dotarły do nas kolejne wspaniałe informacje o zakończonych sukcesem kolejnych zbiórkach dla Julci i Kacperka, którzy podobnie jak Alex mogą udać się do USA, aby skorzystać z terapii genowej.
Firma Avexis ogłosiła, że od początku 2020 roku organizować będzie losowanie darmowych dawek Zolgensmy dla pacjentów cierpiących na SMA. Forma wyboru osób, którzy będą mieli szansę skorzystać z terapii budzi ogromne kontrowersje i spotyka się z ogromną krytyką ze strony pacjentów i lekarzy. Jest to jednak temat tak obszerny, że wymaga on osobnego artykułu. 😉
Na chwilę obecną nie ma wystarczających dowodów by stwierdzić, która z dostępnych terapii jest skuteczniejsza – nusinersen czy terapia genowa. Wiemy natomiast na pewno, że obie terapie przynoszą najlepsze wyniki, jeśli podane są przed wystąpieniem objawów choroby, w pierwszych tygodniach życia dziecka.
Terapie w zaawansowanej fazie rozwoju
Poza opisanymi wyżej lekami istnieją również inne programy badawcze, mające na celu odkrycie skutecznego środka na rdzeniowy zanik mięśni. Niektóre z nich są na początku długiej drogi, inne z kolei osiągnęły już znaczny postęp i są zdecydowanie bliżej etapu rejestracji. Najbardziej zaawansowany etap rozwoju osiągnął doustny lek Risdiplam, który jest w trakcie badań klinicznych dając przy tym bardzo pozytywne wyniki [18]. Lek ten ma postać syropu, a cząsteczki w nim zawarte są szybko dystrybuowane do wszystkich tkanek organizmu – również komórek nerwowych w ośrodkowym układzie nerwowym. Tak jak nusinersen, risdiplam modyfikuje proces produkcji białka z genu SMN2 zwiększając ilość funkcjonalnego białka otrzymanego właśnie z tego genu [9].
Ważna jest jak najwcześniejsza diagnoza – czas to neuron
Wyniki leczenia opisanymi powyżej sposobami wskazują jednoznacznie, że podanie leku daje tym lepsze rezultaty, im wcześniej zostanie on zaaplikowany. Co więcej, działanie leków sprawdzono u dzieci, u których objawy choroby jeszcze nie wystąpiły, ale badania genetyczne jednoznacznie potwierdziły rdzeniowy zanik mięśni. Okazało się, że zarówno Spinraza jak
i Zolgensma podane przedobjawowo noworodkom z SMA powodowały niespotykanie łagodny przebieg choroby lub nawet całkowity brak objawów – znaczna większość dzieci osiągała kolejne etapy rozwoju motorycznego w typowym dla zdrowych dzieci okresie [19].
Niedobór białka SMN w neuronach ruchowych powoduje obumieranie tych komórek. Podanie leku zwiększa ilości funkcjonalnego białka w komórkach nerwowych, jednak tylko tych, które nie uległy jeszcze zbyt dużej degeneracji. Właśnie dlatego fundamentalne jest jak najszybsze rozpoznanie choroby dziecka. Badania przesiewowe noworodków pod katem SMA dają dziś możliwość postawienia odpowiedniej diagnozy i rozpoczęcia terapii nawet przed wystąpieniem pierwszych objawów. Pierwsze programy badań przesiewowych są prowadzone w kilku krajach Europy, a w Polsce trwają intensywne prace nad ich wprowadzeniem.
W Polsce pacjentów chorych na SMA i ich rodziny zrzesza Fundacja SMA, która zajmuje się m.in. przyspieszeniem dostępu do leczenia, szerzeniem wiedzy na temat SMA, pomocą psychologiczną, i integracją.
Źródła:
1. D’Amico, Adele, et al. “Spinal muscular atrophy.” Orphanet journal of rare diseases 6.1 (2011): 1-10.
2. Zerres, Klaus, and Sabine Rudnik-Schöneborn. “Natural history in proximal spinal muscular atrophy: clinical analysis of 445 patients and suggestions for a modification of existing classifications.” Archives of neurology 52.5 (1995): 518-523.
3. Munsat, Theodore L., and Kay E. Davies. “International SMA consortium meeting (26–28 June 1992, Bonn, Germany).” Neuromuscular Disorders 2.5 (1992): 423-428.
4. Finkel, Richard S., et al. “Observational study of spinal muscular atrophy type I and implications for clinical trials.” Neurology 83.9 (2014): 810-817.
5. Prior, Thomas W., et al. “Newborn and carrier screening for spinal muscular atrophy.” American journal of medical genetics Part A 152.7 (2010): 1608-1616.
6. Lefebvre, Suzie, et al. “Identification and characterization of a spinal muscular atrophy-determining gene.” Cell 80.1 (1995): 155-165.
7. Monani, Umrao R., et al. “A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2.” Human molecular genetics 8.7 (1999): 1177-1183.
8. https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy
9. Farrar, Michelle A., et al. “Emerging therapies and challenges in spinal muscular atrophy.” Annals of neurology 81.3 (2017): 355-368.
10. Luu, Kenneth T., et al. “Population pharmacokinetics of nusinersen in the cerebral spinal fluid and plasma of pediatric patients with spinal muscular atrophy following intrathecal administrations.” The Journal of Clinical Pharmacology 57.8 (2017): 1031-1041.
11. https://www.ema.europa.eu/en/medicines/human/EPAR/spinraza
12. https://www.gov.pl/web/zdrowie/leki-na-sma-raka-pluc-i-bialaczke-na-nowej-liscie-refundacyjnej
13. Canadian Agency for Drugs and Technologies in Health. “Pharmacoeconomic Review Report. Nusinersen (Spinraza)(Biogen Canada Inc.).” (2018).
14. https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease
15. Dabbous, Omar, et al. “Survival, motor function, and motor milestones: comparison of AVXS-101 relative to nusinersen for the treatment of infants with spinal muscular atrophy type 1.” Advances in therapy 36.5 (2019): 1164-1176.
16. Mendell, Jerry R., et al. “Single-dose gene-replacement therapy for spinal muscular atrophy.” New England Journal of Medicine 377.18 (2017): 1713-1722.
17. https://www.novartis.com/news/media-releases/avexis-announces-innovative-zolgensma-gene-therapy-access-programs-us-payers-and-families
18. https://www.roche.com/investors/updates/inv-update-2020-01-23.htm
19. Dangouloff, Tamara, and Laurent Servais. “Clinical Evidence Supporting Early Treatment Of Patients With Spinal Muscular Atrophy: Current Perspectives.” Therapeutics and clinical risk management 15 (2019): 1153.




