
Autorki tekstu: Katarzyna Chwalenia, Anna Kordala
Rdzeniowy zanik mięśni (ang. SMA – Spinal muscular atrophy) to choroba genetyczna, na którą w Polsce choruje około 1000 osób. We wcześniejszych wpisach, opisywaliśmy jej podłoże genetyczne oraz dwa z trzech obecnie dostępnych leków – antysensowny oligonukleotyd nusinersen (Spinraza®, IONIS/Biogen) i terapię genową onasemnogen abeprawowek (Zolgensma®, Novartis Gene Therapies). Dzisiaj więcej o trzecim dostępnym leku – risdiplamie (Evrysdi™, Roche i PTC Therapeutics).
Dla przypomnienia, SMA spowodowane jest mutacją homozygotyczną genu SMN1 (ang. survival motor neuron 1) odpowiedzialnego za produkcję białka SMN. Białko to reguluje wiele procesów w różnych komórkach, ale jego brak najbardziej dotyka neurony ruchowe łączące rdzeń kręgowy i pień mózgu z mięśniami 1,2. W konsekwencji, nie otrzymujące sygnałów z układu nerwowego mięśnie z czasem słabną i zanikają. Wiadomo jednak, że SMA uwidacznia się nie tylko w tkance nerwowej czy mięśniowej. Układ sercowo-naczyniowy, rozrodczy, szkieletowy czy pokarmowy u osób chorych również wykazują szereg zaburzeń. Z tego względu, SMA coraz częściej traktuje się jako chorobę ogólnoustrojową, a osoby chore wymagają obserwacji wielu specjalistów 3.
W Polsce co roku rodzi się około 50 dzieci z SMA. Jak najwcześniejsze wdrożenie leczenia jest kluczowe dla jego efektywności i daje szanse na normalny lub prawie normalny rozwój. Od początku 2019 roku, w naszym kraju osoby chore mają darmowy dostęp do nusinersenu. Możliwe, że już niedługo na podobną ścieżkę wkroczą również terapia genowa oraz risdiplam. Różnorodność łatwo dostępnych terapii jest niezwykle istotna z perspektywy pacjentów, ich rodzin i lekarzy. Jak działa trzeci lek na SMA?
Trzeci lek na SMA – Risdiplam
Risdiplam (Evrysdi™) został zatwierdzony przez Amerykańską Agencję Żywności i Leków (FDA; Food and Drug Administration) 7 sierpnia 2020 roku4, a przez Europejską Agencję Leków (EMA; European Medicines Agency) 26 marca 20215.
Podobnie do opisanego wcześniej nusinersenu – risdiplam modyfikuje mRNA genu SMN2. Gen ten jest prawie identyczny jak SMN1. Prawie, bo SMN2 zawiera małą mutację, która powoduje, że tylko 10-15% białka SMN produkowanego z SMN2 jest prawidłowe. Dzieje się tak bo wspomniana mała mutacja prowadzi do „wypadnięcia” części informacji z mRNA (ekson 7) SMN2, w związku z czym powstające białko SMN jest niekompletne i niefunkcjonalne. Osoby chore na SMA mają jedną lub więcej kopii genu SMN2, a strategia naprawy mRNA SMN2 wydaje się być najbardziej powszechną wśród testowanych produktów leczniczych. Z dopuszczonych leków, tylko terapia genowa opiera się o inny mechanizm działania.
Zarówno risdiplam jak i nusinersen można nazwać „molekularnymi plastrami”, które zmuszają komórkę do uwzględnienia kompletnej informacji w mRNA SMN2 i produkcji prawidłowego białka. Skoro działają podobnie to dlaczego oba leki zostały dopuszczone na rynek? Mimo że mechanizm działania jest analogiczny, istnieją diametralne różnice pomiędzy tymi terapeutykami. Po pierwsze, risdiplam należy do grupy leków zwanych małymi cząsteczkami (ang. small molecules) a nusinersen to antysensowny oligonukleotyd (ang. antisense oligonucleotide). Z chemicznego punktu widzenia, są to całkowicie różne leki, a risdiplam jest zdecydowanie mniejszy od nusinersenu. Niesie to za sobą bardzo istotne konsekwencje. Risdiplam, w przeciwieństwie do nusinersenu, bardzo szybko wchłania się do krwi i dalej tkanek, z łatwością pokonuje wiele barier – w tym niezwykle istotną z perspektywy SMA barierę krew-mózg. W ten sposób lek ten jest w stanie efektywnie dotrzeć do neuronów ruchowych, czego nusinersen dokonać nie potrafi. To właśnie dlatego nusinersen podaje się bezpośrednio do kanału kręgowego przez punkcję lędźwiową, a risdiplam można przyjmować w domu w formie smakowego syropu! Z perspektywy pacjenta jest to niezwykle istotna różnica. Ponado risdiplam doprowadza do poniesienia poziomu białka SMN w całym organizmie pacjenta, wszystkich jego tkankach, a nusinersen działa tylko na układ nerwowy pacjenta.
Badania kliniczne
Badania kliniczne nad risdiplamem dalej trwają. Można je bardzo łatwo zidentyfikować, bo biorą swoje nazwy od różnych… ryb: Firefish6, Sunfish7, Jewelfish8, i Rainbowfish9. Ich różnorodność jest imponująca pod wieloma względami. Nie ograniczają się one wyłącznie do maluchów, ale uwzględniają też dorosłych z SMA, osoby leczone wcześniej innymi lekami (terapią genową czy nusinersenem) albo przedobjawowe noworodki do 6-tygodnia życia. Firefish, Sunfish i Jewelfish rozpoczęły się w 2-3 miesięcznych odstępach, pod koniec 2016 i na początku 2017 roku. To właśnie te badania przyczyniły się do wprowadzenia risdiplamu na europejski i amerykański rynek. Wszystkie cztery badania prowadzone są również w Polsce, jednak na chwilę obecną (sierpień 2021) tylko Rainbowfish prowadzi aktywną rekrutację. Podobnie jak w przypadku innych chorób rzadkich, liczba pacjentów biorących udział w badaniach jest niska (odpowiednio: 62, 231, 174 i 25). Nie zaskakujący jest również fakt, że większość tych badań jest otwarta (bez kontroli placebo) z wyjątkiem badania Sunfish, które w części pierwszej trwającej 12 miesięcy posiadało grupę kontrolną otrzymującą placebo. W drugiej części, wszyscy uczestnicy badania otrzymali lek.
Jak zawsze, pierwszym celem badań klinicznych jest określenie profilu bezpieczeństwa leku. Risdiplam jest bezpiecznym, dobrze tolerowanym lekiem, a efekty uboczne obejmują: gorączkę, biegunkę, wysypkę, ból głowy. Rzadziej, obserwuje się ból stawów, zapalenie pęcherza czy nudności. Ciągle napływające wyniki wszystkich badań klinicznych utwierdzają w przekonaniu o bezpieczeństwie i, co ważne, niezwykłej efektywności risdiplamu. W świecie SMA, określenie efektywności leku opiera się na obserwacji i osiąganiu przez pacjentów tzw. kamieni milowych, czyli między innymi: siedzenie bez podparcia, przewracanie się, raczkowanie, stanie bez podparcia, stanie z asystą czy samodzielne chodzenie. Pamiętajmy, że przed wprowadzeniem leczenia, najcięższe przypadki SMA nie pozwalały dziecku na samodzielne przewracanie się czy podnoszenie główki, a SMA było najczęstszą genetyczną przyczyną śmierci przed drugim rokiem życia! Najnowsze wyniki dostarczone z badania Firefish dotyczą najcięższego typu choroby – SMA110. Średni wiek uczestników to 20,5 miesiąca. 95% uczestników ma się dobrze, u 90% zauważono znaczną poprawę funkcji motorycznych, a 85% nie wymaga wspomagania oddechowego. 29% dzieci jest w stanie siedzieć bez wsparcia przynajmniej przez 5 sekund – kamień milowy, który bez leczenia jest nieosiągalny.
Jeden z najważniejszych wniosków, które na przestrzeni lat wysunięto dzięki obserwacjom, badaniom (klinicznym jak i przed-klinicznym) to konieczność jak najwcześniejszego rozpoczęcia leczenia. Takie podejście dotyczy wszystkich dostępnych terapii, których podsumowanie i porównanie przedstawiamy poniżej.
Tabela podsumowująca dostępne terapie – stan na sierpień 2021
| Nazwa handlowa | Zolgensma | Spinraza | Evrysdi |
| Substancja czynna | Onasemnogen abeparwowek | Nusinersen | Risdiplam |
| Budowa chemiczna | niereplikujący rekombinowany wektor oparty na wirusie związanym z adenowirusami serotypu 9 (AAV9) zawierający cDNA ludzkiego genu SMN1 | syntetyczny oligonukleotyd antysensowny (rodzaj materiału genetycznego) | mała cząsteczka (small molecule) |
| Postać Farmaceutyczna | roztwór do infuzji | roztwór do wstrzykiwań | proszek do sporządzania roztworu |
| Droga i sposób podania | wlew dożylny; podanie przez pompę infuzyjną w postaci pojedynczej i powolnej infuzji trwającej około 60 minut | dooponowo przez nakłucie lędźwiowe; wstrzyknięcie dawki leku do dolnej części pleców | doustnie (syrop) |
| Częstotliwość i dawkowanie | jednorazowe podanie jak najwcześniej po rozpoznaniu choroby; ≥ 2,6 kg nie określono bezpieczeństwa ani skuteczności stosowania terapii u dzieci powyżej 2rż lub 13,5kg | wielokrotne podanie jak najwcześniej po rozpoznaniu choroby w dawce 12 mg (5 ml) w pierwszym dniu leczenia (dniu 0), następnie około dnia 14, dnia 28 i dnia 63, następnie raz na 4 miesiące | wielokrotne podanie raz na dobę po posiłku; ≥ 2 mc-a życia do < 2 lat – 0,20 mg/kg mc; ≥ 2 lat (< 20 kg) – 0,25 mg/kg mc; ≥ 2 lat (≥ 20 kg) 5 mg; należy przyjąć bezpośrednio po pobraniu do strzykawki |
| Mechanizm działania | onasemnogen abeparwowek powoduje ekspresję ludzkiego białka SMN dostarczając funkcjonalną kopię genu SMN1 | nusinersen modyfikuje splicing SMN2 doprowadzając do większej produkcji prawidłowego białka SMN; | ridisplam modyfikuje splicing SMN2 doprowadzając do większej produkcji prawidłowego białka SMN; |
| Najczęstsze działania niepożądane | zaburzenia pracy wątroby i zwiększenie aktywności aminotransferaz wątrobowych, gorączka, wymioty, mikroangiopatia zakrzepowa | zaburzenia układu nerwowego (bóle głowy), zaburzenia mięśniowo-szkieletowe i tkanki łącznej (bóle pleców), zaburzenia żołądka i jelit (wymioty); działania niepożądane uznano za związane z nakłuciem lędźwiowym | gorączka, wysypka, biegunka, nudności*, zakażenie układu moczowego, owrzodzenia jamy ustnej i afty, bóle głowy*, bóle stawów* *dotyczy SMA o późniejszym początku niż niemowlęcy |
| Przeciwwskazania | uczulenie na onasemnogen abeparwowek lub którykolwiek z pozostałych składników tego leku; należy zachować ostrożność w przypadku współistniejących chorób wątroby, małopłytkowości, zakażeń układu oddechowego, niedoczynności kory nadnerczy, gdy miano przeciwciał AAV9 wynosi powyżej 1:50 | uczulenie na nusinersen lub którykolwiek z pozostałych składników tego leku; należy zachować ostrożność w przypadku współistniejących chorób związanych z zaburzeniem krzepnięcia krwi, chorobami nerek i chorobami wątroby | uczulenie na risdiplam lub którykolwiek z pozostałych składników tego leku; należy zachować ostrożność w przypadku współistniejących chorób |
| Koszt (USA) | 2,125 mln USD | 750 000 USD w pierwszym roku i 375 000 USD w każdym kolejnym roku terapii | 340 000 USD za rok terapii |
| Rejestracja w UE | 18 maja 2020 | 30 maja 2017 | 30 marca 2021 |
| Refundacja w Polsce | Proces refundacyjny trwa. | Tak | Proces refundacyjny trwa. |
| Lek dodatkowo monitorowany ▼ | Tak | Tak | Tak |
| Podmiot odpowiedzialny | Novartis Gene Therapies Irlandia | Biogen Netherlands BV Holandia | Roche Registration GmbH, Niemcy |
Dlaczego wczesne rozpoczęcie leczenia w SMA jest kluczowe? Po co nam przesiew noworodków?
Jednym słowem – ponieważ im wcześniej rozpoczniemy leczenie, tym lepsze będą jego efekty.
Wynika to z kilku kwestii. Po pierwsze, jak już wiecie, w SMA umierają neurony ruchowe, których żadne obecne leczenie nie jest w stanie przywrócić do życia. Jeżeli leczenie zostanie rozpoczęte wcześniej, to chory ma więcej neuronów ruchowych, które można odratować.
Proces zaniku neuronów rozpoczyna się zanim pojawią się objawy SMA11. Oznacza to, że kiedy rodzice zaczynają obserwować niepokojące objawy u swojego dziecka, bardzo duży procent neuronów ruchowych uległ już bezpowrotnej degradacji – nie da się ich przywrócić. Dlatego najlepszym rozwiązaniem jest rozpoczęcie terapii na podstawie wyników badania genetycznego, przed wystąpieniem objawów. Aby to było możliwe na szeroką skalę, konieczne są powszechne badania wszystkich noworodków pod kątem SMA. Badania te wykonywane są bardzo popularną techniką PCR.
Dla wszystkich trzech dostępnych w Europie terapii toczyły się lub nadal toczą się badania kliniczne, w których leczeniu poddano bezobjawowe noworodki. Nusinersen (Spinraza) – NURTURE, onasemnogen abeprawowek (Zolgensma) – SPR1NT, risdiplam (Evrysdi) – Rainbowfish. Oczywiście pomiędzy badaniami istnieją różnice, ale wyniki wszystkich pokazują, że leczenie rozpoczęte przed wystąpieniem objawów daje rewelacyjne efekty.
Polska rozpoczęła badanie przesiewowe pod kątem SMA w kwietniu 2021 w kilku województwach, przesiew obejmie cały kraj w drugiej połowie 2022 i od tamtej pory każdy noworodek w Polsce będzie przebadany pod kątem SMA (i wielu innych chorób, które od lat są w programie przesiewowym).
Zdiagnozowanie noworodka i rozpoczęcie leczenia w pierwszych tygodniach życia może umożliwić mu normalny lub niemalże normalny rozwój i zapobiegnie ciężkiej i nieodwracalnej niepełnosprawności u około 50 niemowląt każdego roku w samej Polsce. Wprowadzenie SMA do badań przesiewowych jest ogromnym sukcesem i już zdiagnozowano, i rozpoczęto leczenie dwójki noworodków (stan na sierpień 2021)12, które mają teraz szanse rozwijać się jak zdrowi rówieśnicy.
Tajwan jak pierwszy kraj wprowadził powszechne przesiewy na SMA w 2014, zanim dostępne było jakiekolwiek leczenie, ponieważ zaobserwowano, że wcześniejsze wprowadzenie prawidłowej opieki i rehabilitacji dziecka powoduje, że stan chorego pogarsza się wolniej. Coraz więcej krajów zdaje sobie sprawę z szansy, jaką daje dodanie SMA do listy badań przesiewowych, w związku z czym programy ruszyły między innymi w Belgii, Niemczech, USA czy Australii.
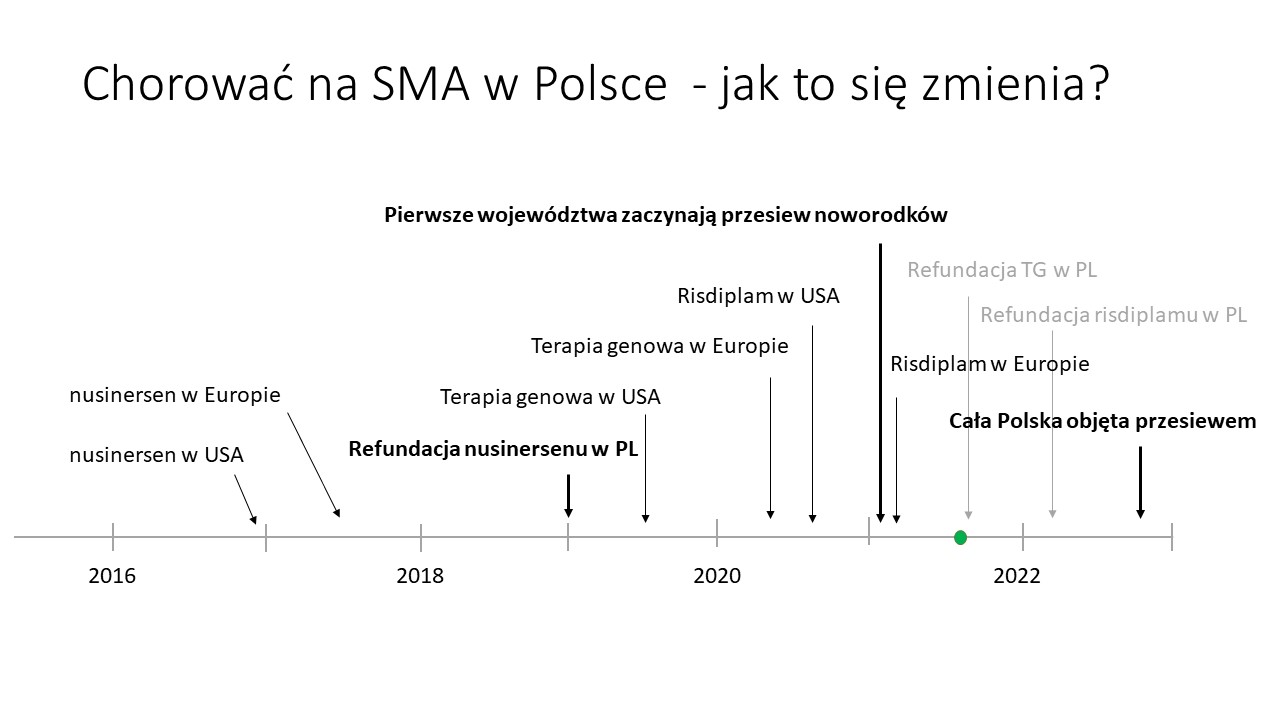
Jak zmieniło się chorowanie na SMA w Polsce?
W przeciągu ostatnich pięciu lat perspektywa życia z SMA przeszła w Polsce i na świecie niesamowitą transformację. Pięć lat temu zatwierdzono w USA pierwszy lek, który od 2019 roku jest refundowany w Polsce. W kolejnych latach zatwierdzono w USA i Europie dwa kolejne – terapię genową Zolgensmę, i risdiplam. W międzyczasie coraz więcej krajów rozpoczęło badania przesiewowe na SMA, w tym Polska. Obecnie mamy sierpień 2021 i dosłownie na dniach zapadnie decyzja o tym, czy Zolgensma będzie w Polsce refundowana i w jakim zakresie. W przyszłym roku zapadnie decyzja o refundacji risdiplamu. Jak widzicie ostatnie lata przyniosły gigantyczne zmiany. Noworodek z SMA urodzony w Polsce jeszcze kilka lat temu nie miał szans na leczenie a jedynie opiekę paliatywną. Noworodek urodzony w 2019 roku miał dostęp do leczenia, ale rozpoczynało się ono po rozpoczęciu objawów i mimo że skuteczne, nie daje szansy na pełną sprawność. I w końcu, dzieci urodzone obecnie mają szanse zostać zdiagnozowane w badaniach przesiewowych, rozpocząć leczenie w pierwszych tygodniach życia i rozwijać się prawidłowo. Mamy nadzieję, że przed nami jeszcze wiele przełomów w dziedzinie chorób rzadkich, ponieważ 95% z nich nadal nie ma zatwierdzonych skutecznych terapii.
Podsumowanie artykułów o SMA
Rdzeniowy zanik mięśni, czyli SMA to genetyczna choroba rzadka, która występuje raz na około 8 000 narodzin. W SMA istnieje poważny niedobór białka SMN, które jest odpowiedzialne za utrzymanie przy życiu neuronów ruchowych. W wyniku choroby neurony zanikają, a mięśnie szkieletowe przez nie unerwiane zaczynają stopniowo zanikać. Najczęściej występuje najostrzejszy typ, typ I, w którym objawy obserwujemy już w pierwszych tygodniach czy miesiącach życia. W ciągu ostatnich kilku lat dopuszczono do użytku trzy terapię – nusinersen, terapię genową i risdiplam. Obecnie (sierpień 2021) w Polsce dla wszystkich chorych refundowany jest nusinersen, podawany punkcją lędźwiową. Toczą się procesy refundacyjne terapii genowej i risdiplamu. Od niedawna kolejne województwa rozpoczynają badanie wszystkich noworodków pod kątem SMA, cała Polska zostanie objęta tymi badaniami pod koniec 2022 roku. Wczesna diagnoza i wprowadzenie leczenia dają szanse zdiagnozowanym dzieciom i ich Rodzinom na normalne, zdrowe życie.
Ten artykuł kończy serię o SMA. W pierwszym omówiliśmy genetykę choroby oraz nusinersen. Drugi był poświęcony terapii genowej, w niniejszym przybliżyłyśmy Czytelnikom risdiplam i wagę wczesnego leczenia. Poza tym można też przeczytać „pigułę” wiedzy o SMA – małe kompendium. Po najnowsze doniesienia i rzetelne materiały zapraszamy na fsma.pl
1. Schrank, B.. et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. U. S. A. 94, 9920–9925 (1997).
2. Groen, E. J. N., Talbot, K. & Gillingwater, T. H. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat. Rev. Neurol. 2018 144 14, 214–224 (2018).
3. Lipnick, SL. et al. Systemic nature of spinal muscular atrophy revealed by studying insurance claims. PLoS One 14, (2019).
4. https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
5. https://www.ema.europa.eu/en/medicines/human/EPAR/evrysdi.
6. https://www.clinicaltrials.gov/ct2/show/NCT02913482.
7. https://www.clinicaltrials.gov/ct2/show/NCT02908685.
8. https://www.clinicaltrials.gov/ct2/show/NCT03032172.
9. https://www.clinicaltrials.gov/ct2/show/NCT03779334.
10. https://www.roche.com/media/releases/med-cor-2021-07-29.htm.
11. Martinez, T. L. et al. Survival Motor Neuron Protein in Motor Neurons Determines Synaptic Integrity in Spinal Muscular Atrophy. J. Neurosci. 32, 8703–8715 (2012).
12. https://www.facebook.com/permalink.php?story_fbid=4812356912127230&id=220646277965006.
Obrazek z Freepik.
Fakty i Mity Genetyki tworzone są przez pasjonatów, specjalistów, naukowców i lekarzy. Ten artykuł czytasz za darmo, bez reklam, bez spamu. Doceń naszą pracę i postaw nam wirtualną kawę 🙂 Dziękujemy! – Wasza Redakcja FiMG





